CEL-Seq is a RNA sequencing technique designed to overcome the limitation of small starting amounts of RNA, often used for single-cell RNA sequencing.
In this protocol, samples are barcoded and pooled before linear amplification of mRNA via IVT.
During the sample preparation, each mRNA transcript is tagged with a Unique Molecular Identifier (UMI), which is used during the bioinformatics analysis to collapse reads originated from the same transcript.
In order to account for pipetting variation, spike-ins of a known concentration may be added to individual samples at the beginning of sample preparation. Data is then mapped and counted according to the known spike-in sequences and annotations. Using the expected counts of spike-ins, correlation plots and statistics are generated.
Researchers who request this service from the TGC receive the following analysis results files:
- Final report – detailed summary of the analysis, including explanations and statistics for each step, technical details, and all necessary information for publishing.
- Raw data – sequenced reads in ‘fastq’ format.
- Genome viewer files – visualization of the aligned reads (bam files), allowing further exploration of specific loci of interest.
- An organized results table – contains normalized expression levels of each gene as well as statistical comparisons tailored to each experiment’s unique requirements.
- Plots – visualization of the data for the researcher’s convenience in the form of a PCA plot, heatmaps, and MA plots.
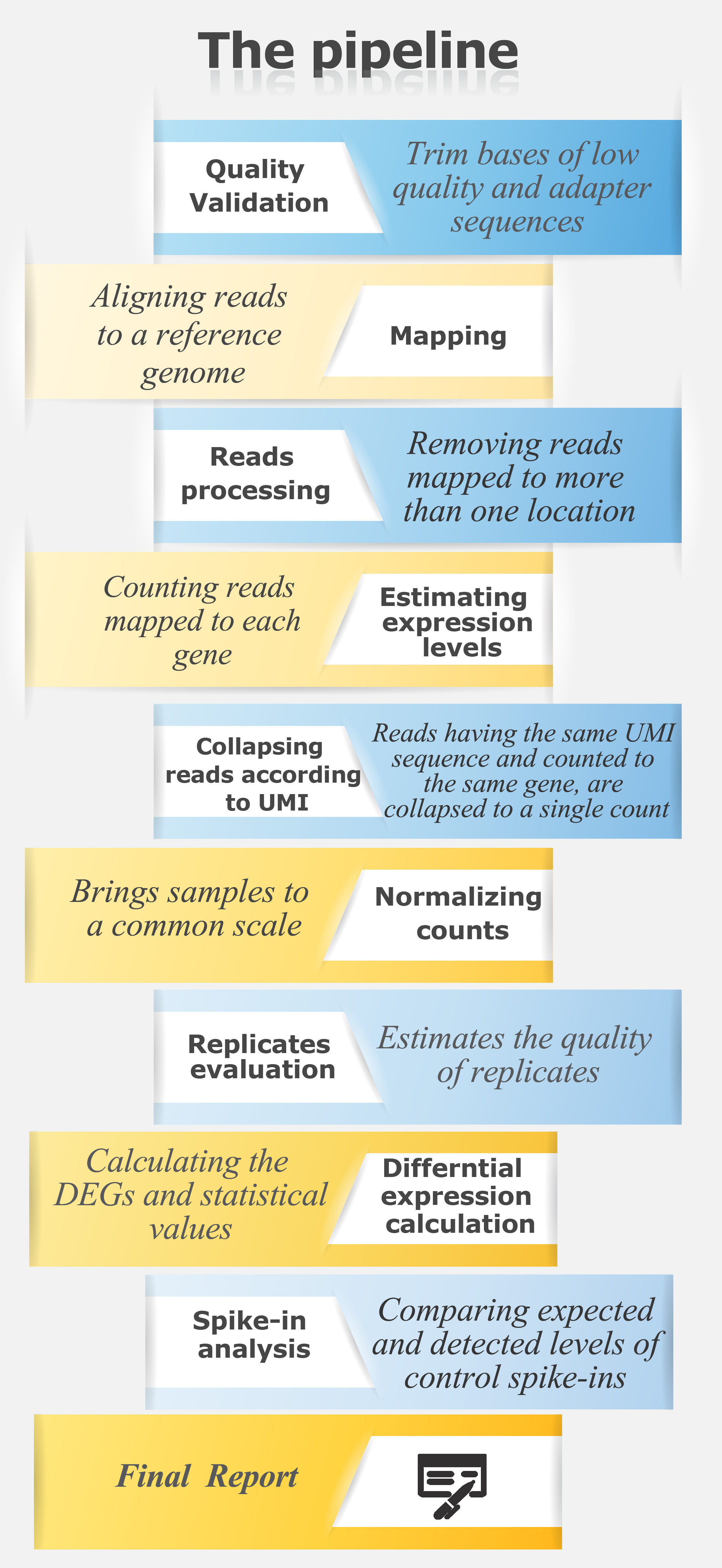
For a CEL-Seq pipeline in text format
Example of Results Table:
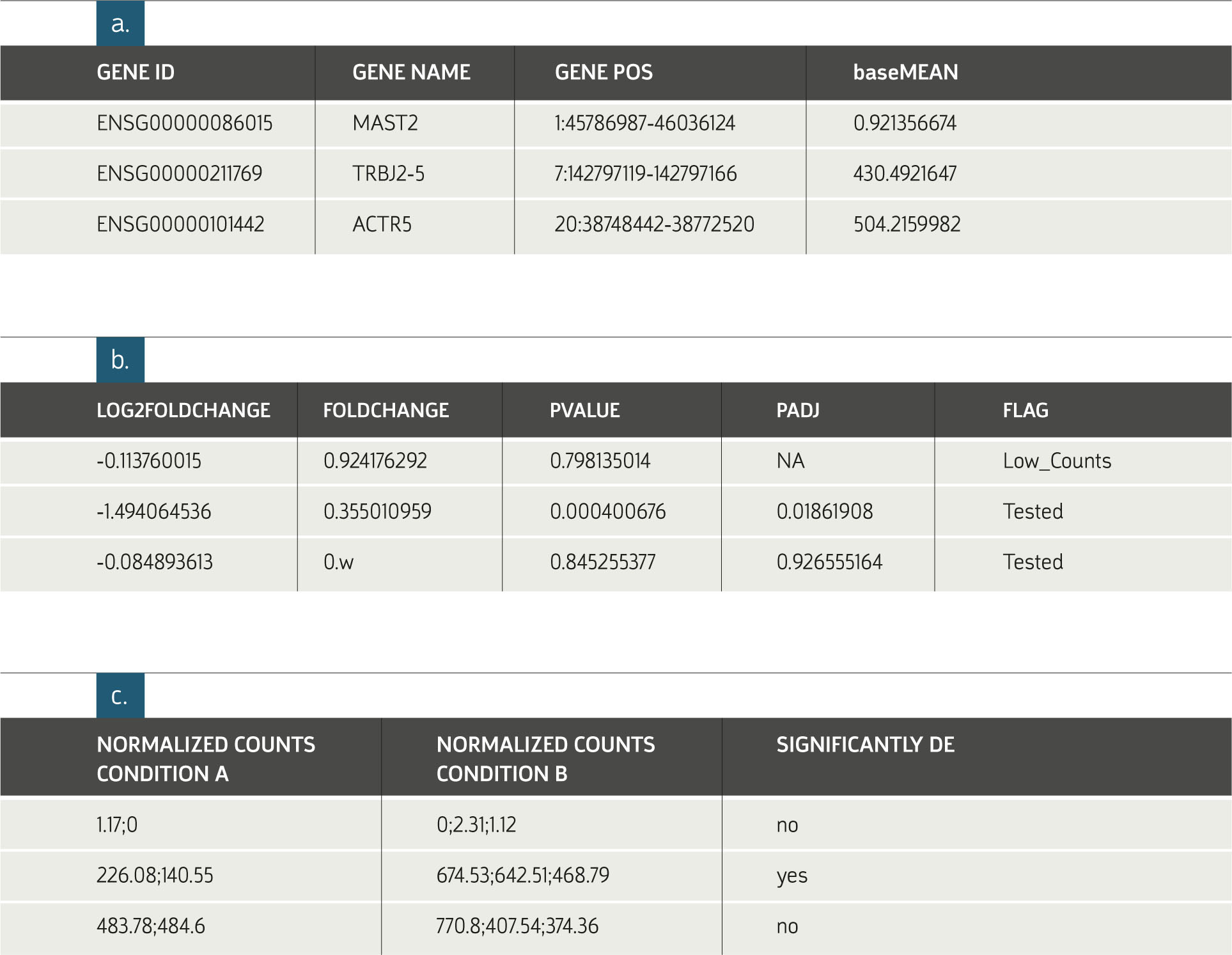
a. General information on each gene. The subjects displayed in this table depend on the organism of interest. The information in figures b. and c. are provided for each requested
comparison between conditions.
b. Statistical results of the differential expression analysis.
c. The normalized expression levels of all samples relevant to each comparison are shown
in columns grouped according to replicate sets. An additional indicator, Significantly DE (Differentially expressed), is provided. This value is based on a minimal threshold on the adjusted p-values.
Examples of CEL-Seq Analysis Plots:

A: PCA plot – PCA analysis is performed in order to visualize and identify experimental covariates and batch effects. The plot spans the samples on a 2-dimensional space demonstrating sample-to-sample distances.
B: Euclidian distances heatmap – This plot allows the researcher to visually quantify similarities and differences between biological replicates and/or different samples under different biological conditions.
C: IGV (genome viewer) –A graphic representation of the mapping patterns of CEL-Seq reads to a reference genome. The example above depicts the mapping of three exonic regions of a single sample, we can see that the mapping pattern shows a clear preference to the 3 prime end of the gene, as expected in CEL-Seq data.